使用Seurat进行聚类和注释
本教程演示如何对预处理的Seurat对象进行聚类,然后使用CASSIA对结果细胞簇进行注释。我们假设质量控制步骤已经完成。
1. 安装和设置
1.1 所需软件包
# 安装所需软件包 install.packages(c("Seurat", "dplyr", "reticulate", "devtools")) # 从GitHub安装CASSIA library(devtools) devtools::install_github("ElliotXie/CASSIA/CASSIA_R") # 加载软件包 library(Seurat) library(dplyr) library(CASSIA) # 设置API密钥(CASSIA运行所需) setLLMApiKey("your_api_key", provider = "openrouter", persist = TRUE) # 或使用anthropic/openaiR
2. 探索预处理的Seurat对象
在本教程中,我们将使用GTEX项目的乳腺组织数据集作为示例。该数据集为乳腺组织细胞类型提供了全面的参考,可从以下链接下载:
# 加载GTEX乳腺数据集 gtex_data <- readRDS("gtex_ref.rds") # 查看数据集元数据 gtex_data@meta.data %>% colnames()R
[1] "orig.ident" "nCount_RNA" "nFeature_RNA" "Broad.cell.type" "Granular.cell.type"
我们可以看到数据集在 Broad.cell.type 和 Granular.cell.type 列中包含金标准细胞类型标签。
3. 降维和聚类
3.1 PCA和UMAP
gtex_data <- NormalizeData(object = gtex_data) gtex_data <- FindVariableFeatures(object = gtex_data) gtex_data <- ScaleData(object = gtex_data) gtex_data <- RunPCA(object = gtex_data) gtex_data <- FindNeighbors(object = gtex_data, dims = 1:25) # 使用25个主成分进行聚类 gtex_data <- FindClusters(object = gtex_data,resolution = 0.4) # 使用分辨率0.4进行聚类 gtex_data <- RunUMAP(object = gtex_data, dims = 1:25) # 使用25个主成分进行UMAP DimPlot(object = gtex_data, reduction = "umap") # 可视化UMAPR
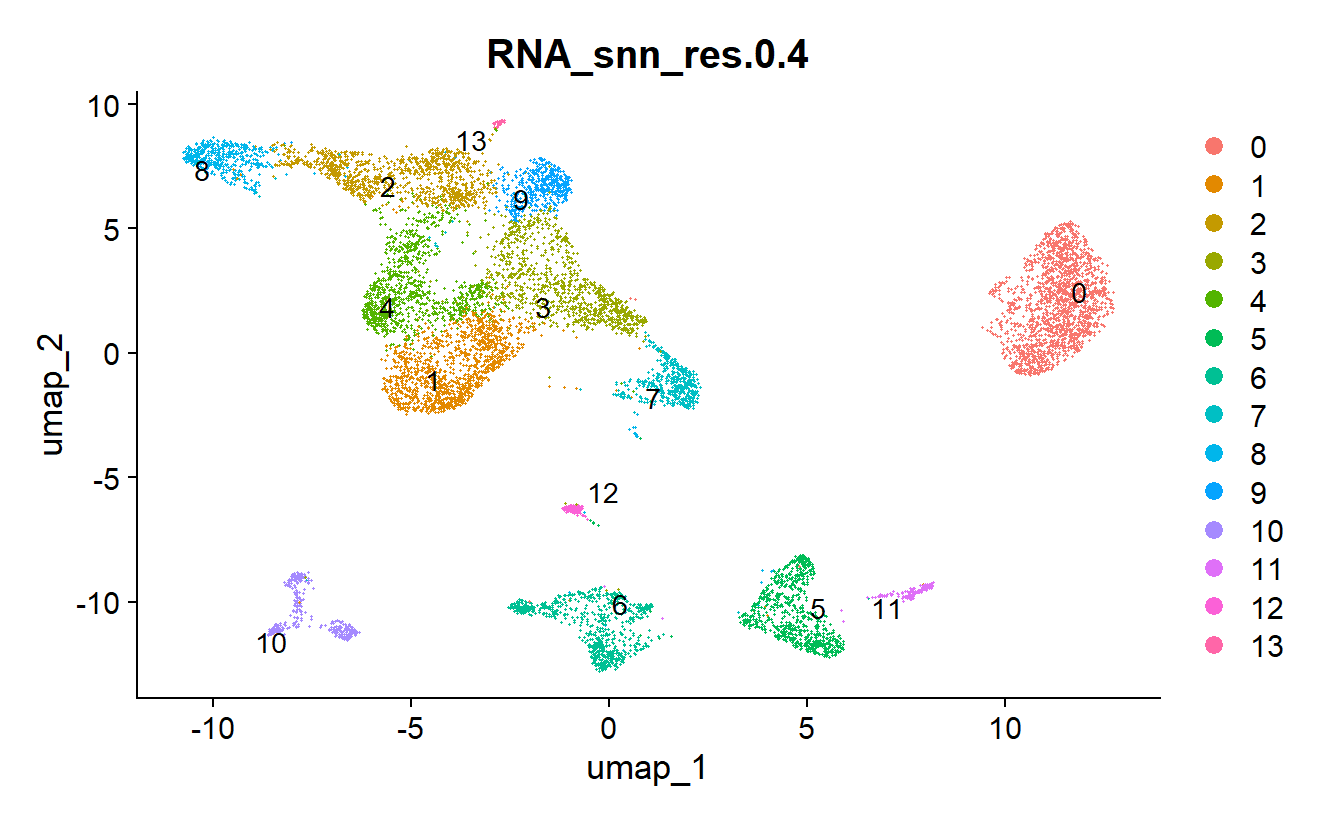
3.3 发现标记基因
Idents(gtex_data)="RNA_snn_res.0.4" # 如果使用默认分辨率0.4,这是默认的细胞簇列名 all_markers <- FindAllMarkers(gtex_data, only.pos = TRUE)R
标记基因过滤在CASSIA内部实现,且注释增强代理也需要原始标记文件;因此,此步骤无需进行过滤。
4. 使用CASSIA注释细胞簇
4.1 基础注释
现在我们有了细胞簇和标记基因,可以使用CASSIA进行注释:
results <- runCASSIA_pipeline( output_file_name = "gtex_breast_annotation", tissue = "Breast", species = "Human", marker = all_markers, max_workers = 6 # 取决于您的CPU核心数 )R
默认提供商是OpenRouter,默认模型已优化以获得最佳注释质量。要查看每个新模型的表现,请访问我们的基准测试网站:
sc-llm-benchmark.pages.dev/methods/cassia
流水线创建一个名为 CASSIA_Pipeline_{tissue}_{species}_{timestamp}/ 的输出文件夹,包含三个子文件夹:
01_annotation_report/- 分析的交互式HTML报告02_annotation_boost/- 低评分细胞簇的注释增强结果03_csv_files/- 汇总CSV文件,包括最终结果
最终结果CSV文件:
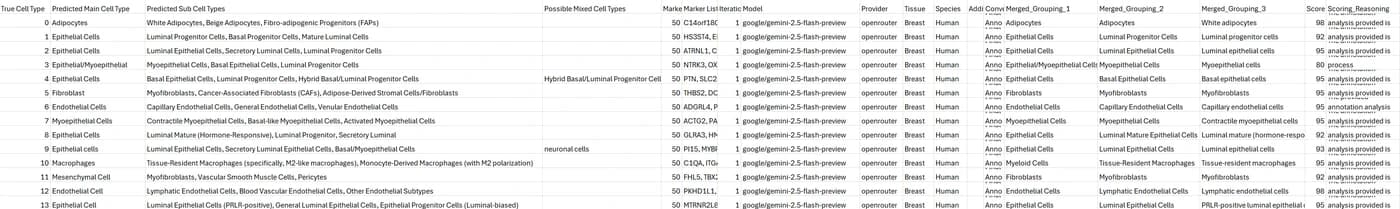
4.2 将注释整合到Seurat对象
运行CASSIA后,您可以将注释整合回Seurat对象:
seurat_breast=add_cassia_to_seurat( seurat_obj = gtex_data, cassia_results_path = "CASSIA_Pipeline_Breast_Human_XXXXXX/03_csv_files/gtex_breast_annotation_FINAL_RESULTS.csv", # 将XXXXXX替换为实际时间戳 cluster_col = "RNA_snn_res.0.4", # Seurat对象中用于FindAllMarkers的细胞簇列名 columns_to_include = 1) # 默认仅导出合并列回seurat,设置为2可导出所有列R
4.3 可视化注释
DimPlot(seurat_breast,group.by = "Broad.cell.type", label = TRUE, repel = TRUE) DimPlot(seurat_breast,group.by = "Granular.cell.type", label = TRUE, repel = TRUE) DimPlot(seurat_breast,group.by = "RNA_snn_res.0.4", label = TRUE, repel = TRUE) DimPlot(seurat_breast,group.by = "CASSIA_merged_grouping_1", label = TRUE, repel = TRUE) DimPlot(seurat_breast,group.by = "CASSIA_merged_grouping_2", label = TRUE, repel = TRUE) DimPlot(seurat_breast,group.by = "CASSIA_merged_grouping_3", label = TRUE, repel = TRUE)R

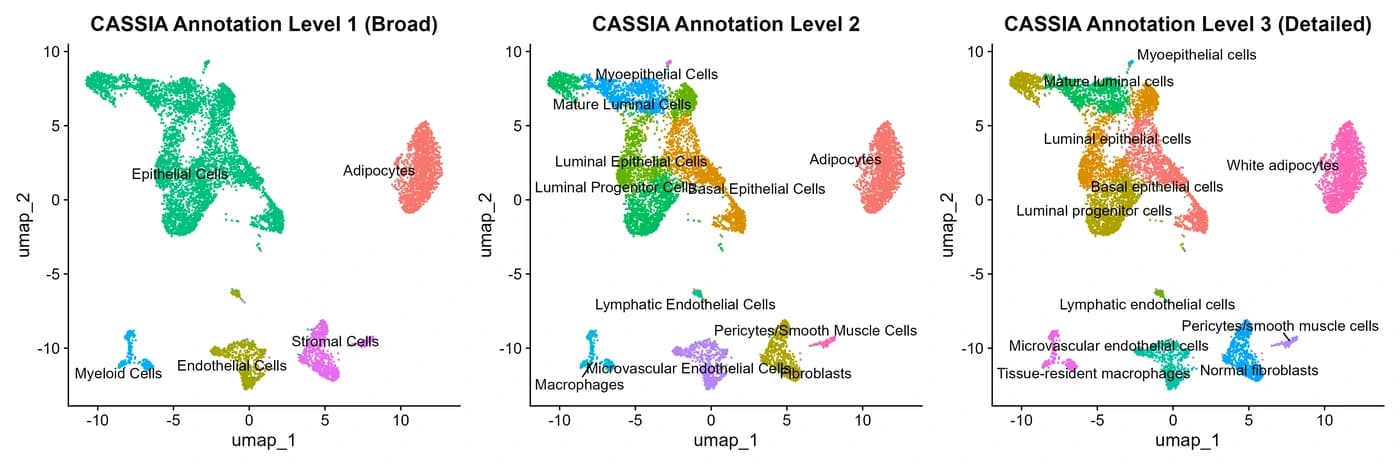
CASSIA会自动将注释汇总为三个不同的详细级别,从最概括到最详细,如UMAP图所示。
5. 后续步骤
完成聚类和注释后:
- 对主要细胞簇进行子集划分并重复上述步骤
- 使用
runCASSIA_annotationboost()处理注释质量较低的细胞簇 - 尝试
compareCelltypes()区分相似的细胞类型 - 探索
runCASSIA_subclusters()对特定细胞群体进行更详细的分析 - 实施
runCASSIA_batch_n_times()进行不确定性量化
有关更全面的质量控制和分析技术,请参阅"完整工作流程最佳实践"教程。